La malattia di Creutzfeldt-Jakob è una malattia neurodegenerativa rara che colpisce rapidamente, progressivamente e gravemente il cervello. Questo disturbo devastante è caratterizzato da una distruzione graduale delle cellule cerebrali, che porta alla formazione di piccoli buchi nel cervello, causando sintomi debilitanti come atassia, andatura anormale, difficoltà nel linguaggio e demenza.
Purtroppo, la CJD è sempre fatale e attualmente non esiste una cura. Ogni anno, a livello globale, si stima che colpisca una persona su un milione, inclusi gli Stati Uniti.
Le cause della CJD possono essere sporadiche, ereditarie o acquisite. Colpisce principalmente le persone di età superiore ai 60 anni, mentre è rara nelle persone di età inferiore ai 30 anni.
È altamente probabile che una persona con CJD non sopravviva oltre un anno dalla comparsa dei sintomi.
Cos’è la CJD?

La CJD è un’encefalopatia spongiforme trasmissibile (TSE) che distrugge il cervello nel tempo. È causata da un agente infettivo noto come prione, che non è un virus né un batterio. Le TSE comprendono anche altre malattie come la sindrome di Gerstmann-Sträussler-Scheinker (GSS), l’insonnia familiare fatale e il kuru negli esseri umani. Altri esempi includono la scrapie nelle pecore e nelle capre e l’encefalopatia spongiforme bovina (BSE), comunemente nota come «malattia della mucca pazza».
Le encefalopatie e le sindromi simili si sono dimostrate trasmissibili in altre specie, e il CDC sottolinea che la CJD classica non è correlata alla BSE o ad altre varianti.
Sintomi
La CJD ha un lungo periodo di incubazione, con sintomi che possono impiegare fino a 40 anni per manifestarsi. La patologia porta a un rapido deterioramento della condizione del paziente, con la maggior parte che muore entro un anno dalla comparsa dei sintomi. I sintomi distintivi includono una rapida progressione verso la demenza e il mioclono, ovvero movimenti spasmodici involontari di gruppi muscolari.
Altri sintomi comuni comprendono cambiamenti di umore e comportamento, alterazioni della personalità, perdita di memoria e compromissione del giudizio. Sebbene la condizione possa assomigliare alla demenza di Alzheimer o alla malattia di Huntington, i sintomi della CJD si sviluppano in giorni o settimane, piuttosto che in anni.
Con il progredire della malattia, i problemi di coordinazione e il mioclono peggiorano, e la vista può deteriorarsi fino alla cecità. Infine, il paziente può perdere la capacità di muoversi o parlare, entrando in coma.
Le autopsie del tessuto cerebrale hanno evidenziato modifiche uniche della CJD che non si riscontrano in altre forme di demenza, e vi sono diverse varianti della CJD con sintomi e decorso della malattia variabili.
Le cause
La CJD si verifica quando una proteina prionica, una forma anomala di proteina amiloide, provoca disfunzioni in altre proteine. L’accumulo e la malformazione dei prioni sulle cellule cerebrali conducono a danni cerebrali e morte. Questa malattia può manifestarsi in forma sporadica, ereditaria o acquisita.
Sporadico CJD
Nell’85% dei casi, la CJD è sporadica, senza evidenti fattori di rischio correlati.
CJD ereditato
Tra il 5 e il 10% dei casi sono ereditati, con mutazioni nel gene responsabile della formazione delle proteine prioniche. Ci possono essere storie familiari di CJD o mutazioni che si verificano nelle cellule uovo o spermatozoi, aumentando il rischio per la prole di sviluppare la malattia.
I prioni non contengono informazioni genetiche e non necessitano di geni per riprodursi, ma una mutazione nel gene della normale proteina prionica può far sì che i prioni agiscano in modo anomalo.
Diverse mutazioni nel gene del prione sono state identificate, e la specifica mutazione presente in ciascuna famiglia influisce sulla frequenza e sui sintomi della malattia. Tuttavia, non tutti coloro che presentano mutazioni nel gene della proteina prionica sviluppano la CJD.
CJD acquisito
Non esistono prove che la CJD possa essere trasmessa da una persona all’altra, ma alcune procedure mediche sono state associate alla trasmissione della malattia. Questi includono:
- Trapianto di cornea
- Impianti di elettrodi
- Innesto della dura madre, noto anche come innesto meningeale
- Uso dell’ormone della crescita umano
Circa l’1% dei casi è trasmesso a seguito di esposizione nota o altamente sospettata al tessuto cerebrale o nervoso compromesso.
Encefalopatia spongiforme bovina
Negli anni ’90, una variante della CJD è stata collegata all’esposizione alla BSE nei bovini. Si credeva che la trasmissione fosse associata al consumo di alimenti contaminati. Questa variante tendeva a colpire pazienti più giovani e mostrava un decorso più lungo.
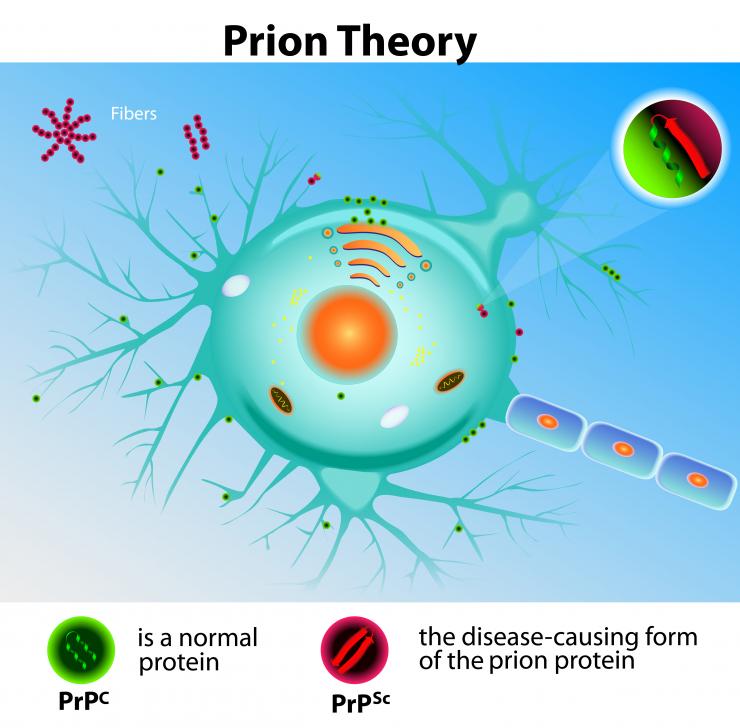
L’ESB colpisce diverse specie, compresi bovini, umani e gatti. Alcuni scienziati ipotizzano che un insolito «virus lento» o un altro organismo possa causare la CJD, ma non sono stati ancora in grado di isolare un virus o un organismo specifico nelle persone affette dalla malattia.
L’agente causale della CJD presenta caratteristiche peculiari non comuni ai virus e ai batteri. Tra queste, un lungo periodo di incubazione, difficoltà di eliminazione e l’assenza di informazioni genetiche sotto forma di acidi nucleici, come DNA o RNA.
Gli scienziati ritengono che la CJD e altre TSE siano causate da prioni, che non sono organismi viventi ma proteine con strutture anomale che si moltiplicano nel cervello, causando danni al tessuto cerebrale e i sintomi caratteristici della malattia.
Diagnosi
Purtroppo, non esiste un test specifico per confermare la diagnosi di CJD. Solo una biopsia cerebrale può fornire una conferma, ma questo procedimento è troppo rischioso per il paziente finché è vivo. Tuttavia, diversi esami possono aiutare a identificare la causa più probabile.
Un esame fisico cercherà spasmi muscolari e valuterà i riflessi del paziente, che possono risultare più reattivi del normale. I muscoli possono essere ipertonici o atonici, a seconda delle aree del cervello colpite.
Esami visivi possono rivelare cecità parziale non riconosciuta dal paziente, mentre un elettroencefalogramma (EEG) può evidenziare impulsi elettrici anormali. TAC o risonanza magnetica possono escludere l’ictus come causa dei sintomi. Inoltre, una puntura lombare può analizzare il liquido spinale per escludere altre cause di demenza, rivelando infezioni o aumento della pressione nel sistema nervoso centrale (SNC).
Se la proteina 14-3-3 viene trovata nel fluido e il paziente presenta sintomi tipici, vi è un’alta probabilità di CJD. Le biopsie cerebrali post-mortem mostrano tessuto cerebrale spugnoso, con piccoli fori visibili dove sono stati distrutti gruppi di cellule nervose.
Trattamento
Sfortunatamente, non esiste una cura per la CJD, e nessun farmaco può rallentare la progressione della malattia. Il trattamento si concentra sull’alleviare i sintomi e sul garantire il massimo comfort al paziente.
Farmaci oppiacei possono essere utilizzati per alleviare il dolore, mentre clonazepam e valproato di sodio possono aiutare a gestire i movimenti involontari. Nelle fasi avanzate della malattia, è fondamentale spostare frequentemente il paziente per prevenire piaghe da decubito, e potrebbe essere necessario un catetere per drenare l’urina, con alimentazione per via endovenosa.
Prevenzione
Le misure preventive includono la sterilizzazione di tutte le attrezzature mediche per eliminare gli organismi potenzialmente responsabili della malattia e la non accettazione di donazioni di cornea da persone con una storia di CJD nota o sospetta.
La maggior parte dei paesi ha ora linee guida rigorose per gestire le mucche infette e restrizioni sui mangimi, al fine di evitare il rischio di trasmissione di altre forme di TSE agli esseri umani.
Si raccomanda a chi è esposto a persone con diagnosi di CJD di seguire alcune linee guida di precauzione, tra cui:
- Copertura di ferite aperte, tagli e abrasioni sulla pelle
- Utilizzo di guanti durante la manipolazione di tessuti, sangue o liquidi del paziente
- Indossare camici monouso o indumenti protettivi in caso di contatto con un paziente
- Utilizzare protezioni per il viso, occhiali o maschere in caso di rischio di spruzzi di liquidi contaminati
- Sterilizzazione dell’attrezzatura utilizzata sopra o vicino al paziente
La ricerca sul ruolo dei prioni nella CJD è in corso, con l’obiettivo di comprendere come la malattia influisca sul cervello e di trovare trattamenti efficaci.
Nuove Ricerche e Prospettive per il 2024
Recenti studi hanno evidenziato l’importanza di approcci innovativi nella diagnosi e nel trattamento della CJD. Nuove tecniche di imaging e biomarcatori nel fluido cerebrospinale stanno emergendo come strumenti promettenti per una diagnosi precoce e più accurata. Inoltre, ricerche in corso si concentrano sull’identificazione di potenziali terapie farmacologiche mirate, che potrebbero rallentare la progressione della malattia.
Una revisione della letteratura scientifica ha rivelato che l’utilizzo di approcci immunologici, come gli anticorpi monoclonali per neutralizzare i prioni, potrebbe rappresentare una frontiera interessante nella lotta contro questa malattia devastante. Anche se attualmente non esistono cure, la comunità scientifica è ottimista riguardo ai progressi futuri e alla possibilità di sviluppare trattamenti efficaci per la CJD.
Inoltre, è fondamentale continuare a sensibilizzare l’opinione pubblica sulla malattia e sull’importanza di segnalare eventuali casi sospetti, poiché una diagnosi e un intervento tempestivi possono fare la differenza nella qualità della vita dei pazienti e delle loro famiglie.