Gli studi clinici sono ricerche fondamentali che hanno lo scopo di stabilire se una strategia medica, un trattamento o un dispositivo è sicuro e efficace per l’uso umano. Questi studi sono essenziali per valutare l’efficacia di approcci medici rivolti a condizioni specifiche o a gruppi di popolazione.
In pratica, le sperimentazioni cliniche contribuiscono all’avanzamento delle conoscenze mediche e forniscono dati affidabili per rafforzare le decisioni e le linee guida nell’assistenza sanitaria.
Per garantire la sicurezza dei partecipanti, le sperimentazioni cominciano con piccoli gruppi, analizzando se un nuovo metodo provoca danni o effetti collaterali indesiderati. È importante notare che un trattamento che ha successo in laboratorio o negli animali potrebbe non essere sicuro o efficace per gli esseri umani.
Fatti veloci sulle sperimentazioni cliniche
- Le prove cliniche mirano a scoprire se una strategia, un trattamento o un dispositivo medico è sicuro ed efficace per l’uso umano.
- Le prove si articolano in quattro fasi e possono concentrarsi su: trattamento, prevenzione, diagnosi, screening, assistenza di supporto, ricerca sui servizi sanitari e scienza di base.
- Un team di ricerca tipicamente include medici, infermieri, assistenti sociali, operatori sanitari, scienziati, responsabili dei dati e coordinatori delle prove cliniche.
- La partecipazione comporta sia rischi che benefici, e i partecipanti devono firmare un documento di «consenso informato» prima di unirsi a uno studio.
- I rischi sono monitorati, ma la natura della ricerca medica implica che alcuni siano inevitabili.
Quali sono gli studi clinici?
Il principale obiettivo delle sperimentazioni cliniche è la ricerca. Questi studi sono progettati per ampliare le conoscenze mediche relative al trattamento, alla diagnosi e alla prevenzione di malattie o condizioni.

Le sperimentazioni seguono rigorosi standard e linee guida scientifiche che mirano a:
- proteggere i partecipanti
- fornire risultati affidabili e precisi
Le prove cliniche sugli esseri umani si svolgono nelle fasi finali di un lungo e sistematico processo di ricerca.
Il processo inizia spesso in laboratorio, dove nuovi concetti vengono sviluppati e testati. I test sugli animali permettono di osservare l’impatto di un approccio su un organismo vivente.
Infine, i test sugli esseri umani vengono condotti in gruppi di dimensioni crescenti.
Le prove possono essere effettuate per:
- Valutare interventi di trattamento per una malattia, una sindrome o una condizione, come farmaci, dispositivi medici o approcci chirurgici e terapeutici
- Valutare modi per prevenire malattie, ad esempio attraverso farmaci, vaccini e modifiche dello stile di vita
- Valutare interventi diagnostici per identificare o diagnosticare specifiche patologie
- Esaminare metodi per riconoscere condizioni o fattori di rischio
- Esplorare procedure di supporto per migliorare il comfort e la qualità della vita dei pazienti con malattie croniche
L’esito di una sperimentazione clinica può rivelare se una nuova strategia, un trattamento o un dispositivo medico:
- ha un effetto positivo sulla prognosi del paziente
- causa danni imprevisti
- non ha benefici positivi o causa effetti negativi
Le sperimentazioni cliniche possono fornire informazioni preziose sull’efficacia dei costi di un trattamento, sul valore clinico di un test diagnostico e su come un trattamento possa migliorare la qualità della vita.
Tipi di sperimentazione clinica
Ogni sperimentazione clinica ha un obiettivo primario, che può essere suddiviso nelle seguenti categorie:
- Trattamento: test di nuovi trattamenti, combinazioni di farmaci o nuovi approcci terapeutici
- Prevenzione: esaminare modi per migliorare la prevenzione o la recidiva della malattia, attraverso medicinali, vitamine, vaccini e cambiamenti dello stile di vita
- Diagnostica: ricerca di tecniche e procedure migliorate per diagnosticare malattie e condizioni
- Screening: test per identificare malattie o condizioni di salute
- Assistenza di supporto: studio di procedure per migliorare il comfort e la qualità della vita dei pazienti con malattie croniche
- Ricerca sui servizi sanitari: valutare l’organizzazione e il finanziamento dell’assistenza sanitaria
- Scienza di base: esaminare il funzionamento di un intervento
Perché gli studi clinici sono importanti?
Le sperimentazioni cliniche sono fondamentali per migliorare e far progredire le cure mediche. Questi studi offrono prove concrete utilizzabili per ottimizzare la cura del paziente.
La ricerca clinica si svolge solo quando ci sono incertezze su:
- se un nuovo approccio è efficace e sicuro per gli esseri umani
- quali trattamenti funzionano meglio per malattie specifiche e gruppi di pazienti
Come funzionano le sperimentazioni cliniche?
Molti elementi sono coinvolti nella creazione, esecuzione e monitoraggio di una sperimentazione clinica.
Protocollo di prove cliniche

Ogni sperimentazione segue un piano o protocollo dettagliato. Questo protocollo è la descrizione scritta di uno studio clinico.
Esso include obiettivi, progettazione, metodi, contesto scientifico e informazioni statistiche rilevanti.
Le informazioni chiave possono includere:
- numero di partecipanti
- criteri di idoneità per la partecipazione
- tipi di test e frequenza di somministrazione
- tipi di dati da raccogliere
- durata dello studio
- piano di trattamento dettagliato
Evitare pregiudizi
I ricercatori devono prendere misure per minimizzare i pregiudizi.
Il bias può derivare da scelte umane o fattori non correlati al protocollo che influenzano i risultati dello studio.
Misure per evitare distorsioni includono l’uso di gruppi di confronto, randomizzazione e mascheramento.
Gruppi di confronto
La maggior parte degli studi clinici utilizza gruppi di confronto per confrontare strategie e trattamenti medici, evidenziando se un gruppo ha un risultato migliore dell’altro.
Questa comparazione avviene in due modi principali:
- Un gruppo riceve un trattamento standard, mentre il secondo gruppo riceve un nuovo trattamento. I ricercatori confrontano poi i risultati ottenuti.
- Un gruppo riceve un nuovo trattamento e il secondo un placebo, un prodotto inattivo simile al trattamento testato.
La randomizzazione
Le sperimentazioni cliniche con gruppi di confronto impiegano frequentemente la randomizzazione. I partecipanti vengono assegnati ai gruppi in modo casuale, garantendo che eventuali differenze riscontrate siano dovute all’intervento e non a differenze preesistenti.
Mascheratura o accecamento
La mascheratura aiuta a prevenire pregiudizi, tenendo ignoti sia i partecipanti che i ricercatori riguardo al trattamento somministrato.
Singolo cieco: quando i partecipanti o i ricercatori non sono a conoscenza del gruppo di assegnazione.
Doppio cieco: entrambi, partecipanti e ricercatori, non sono a conoscenza della assegnazione al gruppo.
Fattori di confondimento
Un confondente può alterare la vera relazione tra due o più variabili.
Ad esempio, si potrebbe erroneamente concludere che le persone che portano un accendino abbiano un rischio più elevato di sviluppare il cancro ai polmoni, quando in realtà il fumo è il fattore di confondimento.
Le persone con un accendino tendono ad essere fumatori, e i fumatori hanno un rischio maggiore di cancro ai polmoni, ma alcune persone possono avere un accendino per altri motivi.
Ignorare questo fattore può portare a conclusioni errate.
Chi è nel team di ricerca?
Ogni studio clinico è guidato da un investigatore principale, generalmente un medico.
Il team di ricerca può includere:
- medici
- infermieri
- lavoratori sociali
- operatori sanitari
- scienziati
- gestori di dati
- coordinatori delle sperimentazioni cliniche
Dove vengono condotti gli studi clinici?
La posizione dipende dal tipo di studio e dall’organizzazione responsabile. Alcuni luoghi comuni includono:
- ospedali
- università
- centri medici
- uffici medici
- cliniche di comunità
- siti di ricerca pubblici e privati
Quanto durano le prove?
La durata varia in base a ciò che viene studiato e ad altri fattori. Alcuni studi durano giorni, mentre altri possono estendersi per anni.
Prima di iscriversi a una sperimentazione, i partecipanti vengono informati della durata prevista.
Progettazione e organizzazione
Esistono diversi tipi di studio e metodi di organizzazione. Di seguito alcuni tipi di studio.
Studi osservazionali
Gli studi di coorte e gli studi caso-controllo sono esempi di studi osservazionali.
Studio di coorte

Uno studio di coorte è un tipo di studio osservazionale in cui è selezionata una popolazione di studio.
Le informazioni sono raccolte per identificare quali soggetti hanno:
- una caratteristica specifica, come un gruppo sanguigno ritenuto correlato allo sviluppo di una malattia
- un’esposizione a un fattore potenzialmente legato a una malattia, come il fumo
Un individuo potrebbe essere selezionato per il suo status di fumatore e poi seguito nel tempo per valutare la probabilità di sviluppare una malattia rispetto ad altri.
Questo tipo di studio è utile per esaminare l’impatto di fattori di rischio che non possono essere controllati sperimentalmente, come il fumo e il cancro al polmone.
I principali vantaggi degli studi di coorte includono:
- L’esposizione viene misurata prima dell’insorgere della malattia, riducendo il rischio di bias.
- Le esposizioni rare possono essere studiate in gruppi adeguati.
- È possibile analizzare più esiti per una singola esposizione.
- Si può calcolare l’incidenza della malattia in gruppi esposti e non esposti.
Tuttavia, ci sono anche svantaggi:
- Possono essere costosi e richiedere tempo, specialmente se condotti prospetticamente.
- Le variazioni temporali dello stato di esposizione e dei criteri diagnostici possono influenzare la classificazione.
- Le perdite al follow-up possono introdurre bias di selezione.
Studi caso-controllo
Uno studio caso-controllo può identificare fattori di rischio per condizioni mediche specifiche.
I ricercatori confrontano le persone con una condizione a quelle senza, risalendo nel tempo per identificare differenze.
Gli studi caso-controllo sono retrospettivi, iniziando dall’esito e indagando le esposizioni.
I principali vantaggi includono:
- I risultati possono essere ottenuti rapidamente.
- Richiedono pochi finanziamenti.
- Sono utili per studiare malattie rare o con lungo periodo di induzione.
- Si può esaminare un’ampia gamma di fattori di rischio.
- Le esposizioni multiple possono essere analizzate.
- Richiedono pochi partecipanti.
Gli svantaggi sono:
- I dati sull’incidenza non possono essere generati.
- Sono soggetti a bias.
- La selezione dei controlli può essere problematica, introducendo bias di selezione.
- Stabilire la sequenza temporale tra esposizione e malattia può essere difficile.
Studio caso-controllo annidato
In uno studio caso-controllo annidato, i gruppi di casi e controlli provengono dalla stessa popolazione di studio.
Durante il follow-up della coorte, i casi emergenti diventano i «casi» dello studio; i partecipanti non interessati diventano «controlli».
Questo approccio è meno costoso e richiede meno tempo rispetto a uno studio di coorte.
I principali vantaggi degli studi caso-controllo nidificati includono:
- Efficienza: non tutti i partecipanti devono essere testati.
- Flessibilità: consentono di testare ipotesi non previste.
- Riduzione del bias di selezione: casi e controlli provengono dalla stessa popolazione.
Tuttavia, i risultati possono avere una validità inferiore a causa delle dimensioni ridotte del campione.
Studio ecologico
Uno studio ecologico esamina la relazione tra esposizione e outcome a livello di popolazione.
Le categorie comuni includono:
- confronti geografici
- analisi temporali
- studi di migrazione
I vantaggi degli studi ecologici comprendono:
- Costi contenuti, poiché utilizzano dati già raccolti.
- Minore dispendio di tempo rispetto ad altri studi.
- Facilità di comprensione.
Tuttavia, presentano svantaggi:
- Possibili errori di deduzione, noti come errori ecologici.
- Difficoltà nel rilevare esposizioni individuali.
- Mancanza di informazioni sui fattori di confondimento.
Studi sperimentali
Oltre agli studi osservazionali, esistono studi sperimentali, inclusi quelli di trattamento.
Studi controllati randomizzati

Uno studio randomizzato controllato (RCT) assegna casualmente i partecipanti a ricevere o meno un intervento specifico.
Possono essere utilizzati due trattamenti diversi o un trattamento e un placebo.
Questo è il tipo di studio più efficace per identificare quale trattamento funziona meglio, riducendo l’influenza di variabili esterne.
I principali vantaggi degli RCT includono:
- Assenza di bias consapevole o inconscio da parte dei ricercatori, garantendo validità esterna.
- Eliminazione delle variabili di confondimento, a patto che il campione sia sufficientemente grande.
Gli svantaggi includono:
- Richiedono tempo.
- Possono essere costosi.
- Richiedono grandi gruppi di partecipanti.
- Eventi rari possono essere difficili da studiare.
Sperimentazione clinica adattativa
Un metodo di progettazione adattativa si basa sui dati raccolti in corso d’opera. È flessibile ed efficiente, consentendo modifiche alle procedure delle sperimentazioni in corso.
Quasi-esperimento
Gli studi quasi sperimentali o «non randomizzati» includono una vasta gamma di interventi non randomizzati, spesso utilizzati quando un RCT non è pratico o etico.
Gerarchia delle prove
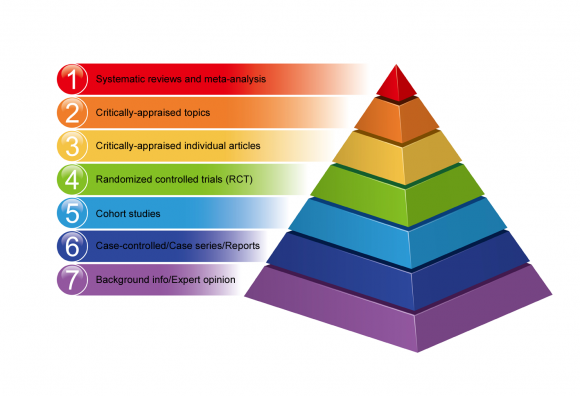
La gerarchia delle prove consente di classificare vari metodi di ricerca in base alla validità delle loro scoperte.
Non tutti i progetti di ricerca sono uguali in termini di rischio di errore e bias. Alcuni metodi forniscono evidenze di maggiore qualità rispetto ad altri.
Di seguito è riportato un esempio di gerarchia della medicina basata sull’evidenza, rappresentata da una piramide, che va da qualità inferiore a qualità superiore delle evidenze.
Fasi di una sperimentazione clinica
Gli studi di ricerca medica sono suddivisi in fasi, definite dalla FDA.
Le prime fasi di sperimentazione indagano la sicurezza di un farmaco e i potenziali effetti collaterali. Le fasi successive testano se un nuovo trattamento è migliore di uno esistente.
Studi di fase 0: farmacodinamica e farmacocinetica
La fase 0 è esplorativa e fornisce informazioni cliniche preliminari su un nuovo farmaco.
Questa fase:
- è condotta all’inizio della fase 1
- comporta esposizione umana molto limitata
- non ha intenti terapeutici, limitandosi a screening e microdosi
Prove di fase 1: screening per la sicurezza
Dopo la fase 0, seguono altre quattro fasi di prove sugli esseri umani, che spesso si sovrappongono. Le fasi da 1 a 3 si svolgono prima della concessione della licenza.
Le linee guida della fase 1 comprendono:
- tra 20 e 80 volontari sani
- verifica degli effetti collaterali più comuni del farmaco
- scoprire come il farmaco viene metabolizzato ed eliminato
Prove di fase 2: stabilire l’efficacia
Se gli studi di fase 1 non mostrano tossicità inaccettabili, iniziano gli studi di fase 2.
Questa fase implica:
- tra 36 e 300 partecipanti
- raccolta di dati preliminari sull’efficacia del farmaco in persone con una specifica condizione
- studi controllati che confrontano chi riceve il farmaco con un gruppo simile che riceve un altro farmaco o un placebo
- continua valutazione della sicurezza
- analisi degli effetti collaterali a breve termine
Prove di fase 3: conferma finale di sicurezza ed efficacia
Se la fase 2 conferma l’efficacia di un farmaco, la FDA e gli sponsor discuteranno come condurre studi su larga scala nella fase 3.
Questa fase comporta:
- tra 300 e 3.000 partecipanti
- raccolta di ulteriori informazioni su sicurezza ed efficacia
- studi su diverse popolazioni
- esame di vari dosaggi per determinare la migliore prescrizione
- uso del farmaco in combinazione con altri per valutarne l’efficacia
Dopo questa fase, le informazioni complete sul nuovo farmaco vengono presentate alle autorità sanitarie.
Riunione riepilogativa
Se la FDA approva il prodotto per il marketing, vengono eseguiti requisiti di post-marketing e studi di impegno.
La FDA utilizza questi studi per raccogliere informazioni sulla sicurezza, efficacia e uso ottimale del prodotto.
Nuova applicazione per i farmaci

Un sponsor di farmaci completa una nuova applicazione per la droga (NDA) per richiedere alla FDA di prendere in considerazione l’approvazione di un nuovo farmaco per la commercializzazione negli Stati Uniti.
Una NDA include:
- tutti i dati animali e umani
- analisi dei dati
- informazioni sul comportamento del farmaco nel corpo
- dettagli di produzione
La FDA ha 60 giorni per decidere se archiviare la richiesta per la revisione.
Se decidono di procedere, un team di revisione esamina la ricerca dello sponsor sulla sicurezza e l’efficacia del farmaco.
I seguenti passaggi devono quindi avvenire.
Etichettatura dei farmaci: la FDA esamina l’etichettatura professionale del farmaco per garantire che le informazioni adeguate vengano comunicate a consumatori e operatori sanitari.
Ispezione della struttura: la FDA controlla i luoghi di produzione del farmaco.
Approvazione del farmaco: i revisori della FDA approvano la domanda o inviano una lettera di risposta.
Prove di fase 4: studi durante le vendite
Le prove di fase 4 si svolgono dopo che il farmaco è stato approvato per la commercializzazione e hanno lo scopo di includere:
- oltre 1.000 pazienti
- un’esperienza completa nella valutazione della sicurezza e dell’efficacia della nuova terapia in una popolazione più ampia e in sottogruppi di pazienti
- confronto e combinazione con altri trattamenti disponibili
- valutazione degli effetti collaterali a lungo termine del farmaco
- rilevamento di eventi avversi meno comuni
- analisi costo-efficacia rispetto ad altre terapie
Rapporto di sicurezza
Dopo l’approvazione della FDA, inizia la fase di post-marketing. Lo sponsor, di solito il produttore, invia periodicamente aggiornamenti di sicurezza alla FDA.
Chi sponsorizza le sperimentazioni cliniche?
Le prove cliniche possono costare centinaia di milioni di dollari. I gruppi che finanziano le sperimentazioni possono includere:
- aziende farmaceutiche, biotecnologiche e di dispositivi medici
- centri medici accademici
- fondazioni volontarie
- l’Istituto Nazionale della Salute
- dipartimenti governativi
- medici e operatori sanitari
- individui
Chi può partecipare?
Il protocollo definisce chi è idoneo a partecipare a una sperimentazione clinica.
I possibili criteri di inclusione possono includere:
- diagnosi di una malattia o condizione specifica
- essere «sani», senza condizioni di salute preesistenti
I criteri di esclusione sono fattori che possono escludere alcune persone dalla partecipazione.
Esempi includono età, sesso, tipo specifico o stadio di malattia, precedenti trattamenti e altre condizioni mediche.
Possibili benefici e rischi
Partecipare a studi clinici può comportare benefici e rischi per i partecipanti.
I potenziali benefici includono:
- Accesso a nuovi trattamenti.
- Se il trattamento risulta efficace, i partecipanti saranno tra i primi a beneficiarne.
- I partecipanti non assegnati al nuovo trattamento possono ricevere il trattamento standard, che può essere uguale o migliore.
- Monitoraggio della salute da parte di un team di professionisti.
- Contributo alla crescita delle conoscenze scientifiche, aiutando altri e migliorando l’assistenza sanitaria.
I potenziali rischi includono:
- Il trattamento standard potrebbe essere superiore ai nuovi approcci studiati.
- Un nuovo trattamento può funzionare per alcuni ma non per altri.
- Effetti collaterali imprevisti, specialmente negli studi di fase 1 e 2.
- Le assicurazioni sanitarie potrebbero non coprire i costi per i pazienti in sperimentazione.
Cosa significa dare il consenso?

Il documento di consenso informato spiega rischi e benefici della partecipazione a uno studio clinico.
Gli elementi chiave nel documento includono:
- scopo della ricerca
- rischi prevedibili di disagio
- possibili benefici
I partecipanti devono leggere attentamente il documento, decidere se iscriversi e firmare per partecipare.
Gli studi clinici sono sicuri?
La FDA si impegna a garantire che chiunque consideri di partecipare a uno studio clinico abbia accesso a informazioni affidabili per prendere decisioni informate, compresi i rischi.
Sebbene i rischi siano controllati e monitorati, alcuni possono essere inevitabili data la natura della ricerca medica.
Come sono protetti i partecipanti?

La sicurezza dei partecipanti è una priorità. Ogni prova è soggetta a supervisione scientifica e tutela dei diritti dei pazienti.
La buona pratica clinica (GCP) mira a garantire che siano seguite procedure etiche e appropriate.
La conformità alla GCP assicura al pubblico che la sicurezza e i diritti dei partecipanti sono protetti.
Le fondamenta della GCP furono stabilite nel 1947, stabilendo che durante le prove i ricercatori devono garantire:
- partecipazione volontaria
- consenso informato
- minimizzazione del rischio
Nel tempo, sono state aggiunte ulteriori misure per proteggere le popolazioni vulnerabili e fornire orientamenti ai ricercatori.
Diritti dei pazienti
I diritti dei pazienti sono protetti attraverso:
Il consenso informato è il processo di fornire ai partecipanti tutti i dettagli relativi allo studio prima della partecipazione. Include informazioni sui trattamenti e test che possono essere ricevuti, oltre a potenziali benefici e rischi.
Altri diritti: il consenso informato non è un contratto vincolante; i partecipanti possono ritirarsi dallo studio in qualsiasi momento.
Diritti e protezione per i minori: un genitore o tutore deve fornire il consenso legale per minori di 18 anni. Se un trattamento comporta rischi elevati, entrambi i genitori devono dare il consenso. I bambini di età superiore ai 7 anni devono acconsentire a partecipare agli studi clinici.
Come trovo una sperimentazione clinica?
Informazioni sulle attuali sperimentazioni cliniche sono disponibili qui.
Ricerca recente e aggiornamenti nel campo delle sperimentazioni cliniche
Nel 2024, le sperimentazioni cliniche continuano a evolversi con nuove tecnologie e metodologie. Recenti studi hanno dimostrato che l’uso di intelligenza artificiale nella selezione dei partecipanti e nell’analisi dei dati può migliorare notevolmente l’efficacia delle prove cliniche. Secondo una ricerca pubblicata nel 2023, l’implementazione di algoritmi predittivi ha portato a una riduzione del 30% nei tempi di reclutamento dei partecipanti, consentendo risultati più rapidi e efficienti.
Inoltre, con l’aumento dell’uso di terapie personalizzate, le sperimentazioni cliniche si stanno adattando per includere approcci più mirati, come le terapie geniche e cellulari. Queste innovazioni richiedono un rigoroso monitoraggio e una valutazione continua per garantire la sicurezza e l’efficacia dei nuovi trattamenti.
Infine, i recenti sviluppi nella comunicazione dei risultati delle sperimentazioni cliniche stanno contribuendo a una maggiore trasparenza. Le piattaforme online ora consentono ai partecipanti e al pubblico di accedere facilmente ai risultati delle ricerche, promuovendo una maggiore consapevolezza e fiducia nelle sperimentazioni cliniche.