La neurofibromatosi è una malattia genetica incurabile del sistema nervoso, che influisce principalmente sullo sviluppo dei tessuti delle cellule nervose. I tumori noti come neurofibromi si sviluppano sui nervi e possono portare a una serie di altri problemi.
Questi tumori possono essere innocui, ma in alcuni casi possono comprimere i nervi e altri tessuti, causando gravi danni. La neurofibromatosi (Nf) rappresenta la più comune malattia neurologica genetica causata da un singolo gene. La mutazione di questo gene comporta un controllo inadeguato del tessuto nervoso.
Esistono tre tipi di neurofibromatosi: Nf1, Nf2 e schwannomatosi, ognuno con caratteristiche specifiche. Questo articolo si concentrerà principalmente su Nf1 e Nf2.
Trattamento

Sebbene non esista una cura definitiva per la neurofibromatosi, i suoi sintomi possono essere gestiti.
I neurofibromi non sono generalmente dolorosi, ma se la loro posizione provoca fastidio, come nel caso in cui sfregano contro vestiti o altre superfici, possono essere rimossi. Questo intervento riduce il rischio di prurito, infezione, intorpidimento e disagio generale, anche se è possibile che ricrescano.
L’ipertensione può essere trattata con farmaci e modifiche dello stile di vita; si raccomandano controlli regolari per monitorare la situazione.
Se i tumori che si sviluppano sul nervo ottico influenzano la vista, la rimozione chirurgica può essere necessaria.
In caso di scoliosi, la curvatura della colonna vertebrale può essere corretta chirurgicamente o mediante l’uso di un tutore. Normalmente, i tumori vengono monitorati regolarmente e il trattamento viene fornito secondo necessità.
Neuroma acustico
La chirurgia per i neuromi acustici non sempre migliora l’udito e, in alcuni casi, può addirittura peggiorarlo. La decisione di rimuovere un neuroma acustico dipende non solo dalla perdita dell’udito, ma anche dalle dimensioni del tumore e dalla sua velocità di crescita.
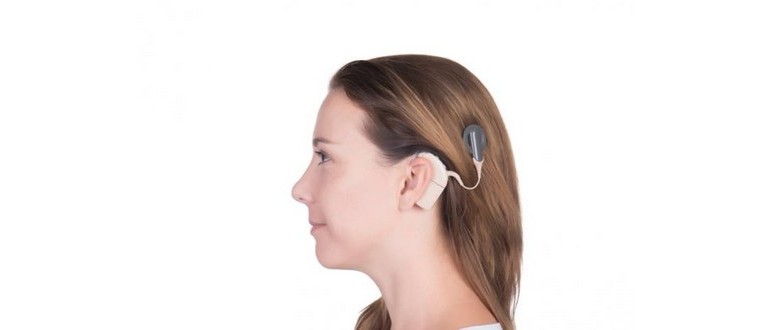
In alcuni casi, un chirurgo può inserire un impianto staminale cerebrale uditivo (ABI) per migliorare l’udito della persona. Questo può avvenire in concomitanza con la rimozione del tumore del nervo vestibolococleare.
Il chirurgo praticherà un’incisione sulla pelle del lato della testa e rimuoverà parte dell’osso dietro l’orecchio per accedere al tumore. Questa procedura consente la rimozione del tumore e l’accesso al tronco cerebrale sottostante.
Le persone con un ABI indossano un ricevitore esterno e un processore vocale, che converte il suono in segnali elettrici inviati all’impianto.
Un impianto cocleare può essere applicato anche dopo la rimozione del tumore. Questo dispositivo elettronico è progettato per fornire una percezione del suono a chi ha problemi di udito profondi o gravi.
Una parte esterna viene posizionata dietro l’orecchio e una seconda parte viene collocata chirurgicamente sotto la pelle. L’impianto include un microfono, un elaboratore vocale e un trasmettitore, che riceve segnali dal processore vocale e li converte in impulsi elettrici.
Un gruppo di elettrodi raccoglie questi impulsi e li invia a varie aree del nervo uditivo.
Sebbene l’impianto non possa ripristinare l’udito normale, può fornire un’utile rappresentazione dei suoni, facilitando la comprensione del linguaggio.
Un audiologo può offrire consigli su come gestire l’equilibrio e l’acufene. Alcune persone con neurofibromatosi imparano a leggere le labbra e utilizzano il linguaggio dei segni.
La radioterapia o la chemioterapia possono aiutare a ridurre i tumori, e attualmente sono in corso ricerche per sviluppare farmaci in grado di prevenire o ridurre la formazione di tumori.
Sintomi
I sintomi della neurofibromatosi variano a seconda del tipo.
Il disturbo può manifestarsi in tutto il corpo, causando tumori e una pigmentazione cutanea anomala. Può presentarsi sotto forma di protuberanze sotto la pelle, macchie colorate, problemi ossei, pressione sulle radici del nervo spinale e altri problemi neurologici.
Inoltre, possono insorgere difficoltà di apprendimento, problemi comportamentali e perdita della vista o dell’udito.
Nf1
Alcune persone con Nf1 presentano solo manifestazioni cutanee e nessun altro problema medico correlato. Segni e sintomi di solito compaiono durante la prima infanzia e, generalmente, non sono dannosi per la salute.
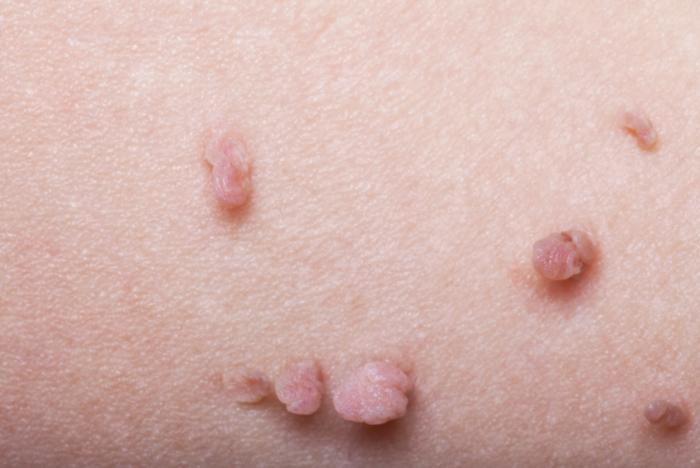
Segni e lentiggini sono comuni; le macchie color caffè possono apparire sulla pelle, spesso fin dalla nascita. Se compaiono più di sei segni all’età di 5 anni, questo potrebbe indicare Nf1. Il numero di macchie può aumentare e col tempo possono diventare più grandi e scure.
Le lentiggini possono apparire in posizioni insolite, come l’inguine, sotto il seno o sotto le ascelle.
I neurofibromi sono tumori, generalmente benigni, che si sviluppano sui nervi della pelle e talvolta anche sui nervi più profondi del corpo, presentandosi come grumi sotto la pelle. Con il passare del tempo, potrebbero svilupparsene di nuovi e potrebbero crescere in dimensioni. I neurofibromi possono apparire molli o sodi e rotondi.
Possono anche verificarsi noduli di Lisch, piccole macchie marroni nell’iride dell’occhio.
Le persone affette da NF1 presentano un rischio maggiore di ipertensione.
NF2
La Nf2 è una condizione più grave in cui i tumori crescono sui nervi più profondi del corpo.
Un neuroma acustico è un tipo comune di tumore cerebrale che si sviluppa sul nervo che collega il cervello all’orecchio interno.
I sintomi possono includere:
- intorpidimento facciale, debolezza e talvolta paralisi
- perdita dell’udito graduale, o più raramente improvvisa
- perdita di equilibrio, vertigini e sensazione di instabilità
- tinnito o ronzio nell’orecchio interessato
Con la crescita del tumore, i sintomi possono peggiorare. Il neuroma può comprimere il tronco cerebrale, il che può risultare pericoloso per la vita. Un piccolo tumore potrebbe non causare alcun problema.
In alcuni casi, i tumori si sviluppano sulla pelle, sul cervello e sul midollo spinale, portando a conseguenze potenzialmente gravi. Alcuni tumori crescono rapidamente, mentre la maggior parte cresce lentamente, rendendo a volte difficile notare l’effetto per diversi anni.
Il monitoraggio regolare permette di rimuovere i tumori prima che insorgano complicazioni.
Possono verificarsi macchie di pigmentazione marrone chiaro, ma saranno meno comuni e numerose rispetto a quelle osservate in Nf1.
Inoltre, possono manifestarsi cataratte; se un bambino sviluppa una cataratta, questo può essere un segno di Nf2. Fortunatamente, questi possono essere facilmente rimossi e generalmente non rappresentano un problema se trattati in tempo.
Tipi
I tre tipi di neurofibromatosi sono Nf1, Nf2 e schwannomatosi.
Neurofibromatosi di tipo 1 (Nf1)
Conosciuta anche come malattia di von Recklinghausen, la neurofibromatosi di tipo 1 è il tipo più comune. La condizione deriva principalmente da una mutazione, piuttosto che da una cancellazione, del gene Nf1, e si stima colpisca 1 persona ogni 3.000.
I segni di nascita possono apparire in diverse parti del corpo poco dopo la nascita.
Durante la tarda infanzia, lesioni o tumori possono comparire sopra o sotto la pelle, variando da pochi a migliaia. In alcuni casi, i tumori possono diventare cancerogeni.
Nf1 può essere a malapena visibile, può apparire antiestetica o può portare a complicazioni potenzialmente gravi; circa il 60% dei casi presenta manifestazioni lievi.
L’uomo elefante ha Nf1?
È un comune malinteso che Joseph Merrick, l’ «Elephant Man», reso famoso dal film del 1980 interpretato da John Hurt, avesse Nf1. Tuttavia, nel 1986, i genetisti hanno dimostrato che Merrick non aveva la sindrome Nf1, ma Proteus, una condizione rara che colpisce meno di 1 persona su 1 milione.
I ricercatori avvertono che confondere le due condizioni potrebbe essere dannoso per le persone con Nf1. In uno studio pubblicato nel 2011, Claire-Marie Legendre e coautori hanno dichiarato:
«Confondere la NF1 con la condizione di Elephant Man danneggia gli interessi di chi ha NF1, in quanto è noto che i pazienti affetti da NF1 hanno difficoltà a stabilire legami sociali e sviluppare una buona autostima».
Gli autori chiedono cambiamenti nell’atteggiamento per dissipare la confusione.
Neurofibromatosi di tipo 2 (Nf2)
La neurofibromatosi bilaterale, o Nf2, deriva normalmente da una mutazione piuttosto che da una delezione del gene Nf2. Questo gene si trova su un cromosoma diverso rispetto a Nf1.
I tumori si formano nel sistema nervoso, solitamente all’interno del cranio, e sono noti come tumori intracranici. Tumori intraspinali possono svilupparsi anche nel canale spinale.
I neuromi acustici sono comuni in Nf2, formando sul nervo vestibolococleare, noto anche come ottavo nervo cranico.
Questo nervo è responsabile dell’udito e influisce sul senso dell’equilibrio; pertanto, può verificarsi perdita dell’udito e dell’equilibrio.
I sintomi tendono a manifestarsi durante la tarda adolescenza e all’inizio degli anni venti, e i tumori possono diventare cancerogeni.
Schwannomatosi
La schwannomatosi è una rara forma di neurofibromatosi geneticamente distinta da Nf1 e Nf2, colpendo meno di 1 persona su 40.000.
Gli schwannomi, o tumori nel tessuto attorno a un nervo, possono svilupparsi in qualsiasi parte del corpo, ad eccezione del nervo vestibolococleare. A differenza di Nf1 e Nf2, non coinvolge i neurofibromi.
I tumori possono causare forte dolore, intorpidimento, formicolio e debolezza nelle dita delle mani e dei piedi.
Le cause

La neurofibromatosi può interessare tutte le cellule della cresta neurale, comprese le cellule di Schwann, i melanociti e i fibroblasti endoneurici, e può influenzare le ossa, causando forti dolori.
Una mutazione di un gene noto come Nf1 è responsabile della Nf1. Questo gene produce normalmente una proteina che regola la crescita del tessuto nervoso.
Tuttavia, nelle persone affette da questa condizione, il gene produce una proteina incompleta, che risulta molto meno efficace nel moderare la crescita dei tessuti nel sistema nervoso, portando così alla formazione di tumori.
Questo gene si trova sul cromosoma numero 17, mentre Nf2 colpisce un gene simile sul cromosoma numero 22.
In circa la metà di tutti i casi noti, entrambi i tipi principali vengono trasmessi da un genitore al figlio, e basta che un genitore abbia il gene difettoso affinché il bambino affronti il rischio di sviluppare neurofibromatosi.
Nell’altra metà dei casi, questi geni subiranno quella che è nota come una mutazione sporadica in uno spermatozoo o in una cellula uovo. Le cause e i fattori di rischio delle mutazioni sporadiche non sono chiari.
Per quanto riguarda Nf2, la mutazione può anche verificarsi dopo la concezione di un embrione in una forma chiamata mosaic Nf2, che è una forma più lieve della malattia.
Diagnosi
La diagnosi di Nf1 avviene normalmente durante l’infanzia. Una diagnosi è confermata quando una persona presenta almeno due dei seguenti segni:
- una storia familiare di Nf1
- un glioma, o tumore, sul nervo ottico, di solito senza sintomi
- lesioni ossee
- almeno sei macchie «café-au-lait» che misurano più di 5 millimetri nei bambini o 15 millimetri negli adolescenti e negli adulti
- almeno due noduli di Lisch o piccole macchie marroni nell’iride
- lentiggini sotto le ascelle, sotto il seno o nella zona inguinale
- due o più neurofibromi o uno «plessiforme»
I neurofibromi plessiformi colpiscono circa il 25% delle persone con Nf1. Questi sono neurofibromi che si diffondono intorno ai grandi nervi man mano che crescono, provocando un ispessimento e una deformità del nervo. Si presentano come nodi o corde sotto la pelle, e possono essere grandi, dolorosi e deturpanti. I plessiformi iniziano normalmente a formarsi durante l’infanzia.
Per la diagnosi, viene utilizzata una lampada speciale per verificare la presenza di segni cutanei. Altri strumenti diagnostici includono una scansione a raggi X, CT o MRI, e può essere usato anche un test del sangue genetico.
Diagnosi di Nf2
I sintomi di Nf2 normalmente compaiono intorno alla pubertà o nell’età adulta, con un’età di insorgenza più comune tra i 18 e i 24 anni.
Una diagnosi di Nf2 è confermata quando si riscontrano:
- neuroma acustico in un orecchio, più due o più sintomi tipici, come cataratta, tumori cerebrali e una storia familiare della condizione
- neuroma acustico in entrambe le orecchie
- neuroma acustico più tumori cerebrali o spinali, rilevati da una risonanza magnetica o da una TAC
- un gene difettoso, identificato attraverso un esame del sangue
Una persona con Nf2 sarà indirizzata a un neurologo, e esami dell’udito e dell’occhio possono essere effettuati per controllare la cataratta, altri problemi oculari e disturbi dell’udito.
Complicazioni
Le complicanze associate a ciascun tipo di neurofibromatosi variano.
Complicazioni di Nf1
Problemi di vista e udito possono insorgere se un tumore o un neuroma preme sui nervi che portano alle orecchie o agli occhi. Di conseguenza, i bambini possono affrontare difficoltà di apprendimento e problematiche comportamentali.
Circa il 50% dei bambini con Nf1 presenta difficoltà di apprendimento. Anche la memoria a breve termine, la consapevolezza spaziale e la coordinazione possono risentirne.
Altre complicazioni di Nf1 includono:
- epilessia
- grande dimensione della testa
- tumori cutanei benigni che, in alcuni casi, possono diventare cancerogeni
- curvatura della colonna vertebrale, o scoliosi
- gliomi, o tumori ai nervi degli occhi, che possono occasionalmente causare problemi alla vista
- ipertensione
- disturbi del linguaggio
- bassa statura
- problemi scheletrici
Possono verificarsi anche sviluppi sessuali precoci o tardivi.
Complicazioni di Nf2
Le persone con NF2 possono sviluppare tumori cutanei benigni, simili a quelli caratteristici di NF1, che devono essere monitorati nel caso in cui crescano, cambino o provochino dolore.
I tumori benigni del cervello possono esercitare pressione su parti del cervello, causando convulsioni, problemi di vista e disturbi dell’equilibrio. Alcuni possono diventare cancerosi.
I tumori del midollo spinale possono svilupparsi sui nervi che circondano la colonna vertebrale, provocando tremori, intorpidimento e dolore agli arti. Tumori nell’area del collo possono causare problemi al viso, come difficoltà nel sorridere, nel battere le palpebre o nella deglutizione.
Prospettive
Molte persone con Nf1 hanno solo sintomi lievi e possono vivere una vita normale e sana. Le complicazioni possono ridurre la durata della vita, ma una persona con Nf1 può spesso aspettarsi di vivere quanto una persona senza la condizione.
Tuttavia, i sintomi richiedono un monitoraggio regolare per prevenire lo sviluppo di complicazioni.
Per quanto riguarda Nf2, la prospettiva è meno favorevole. Anche se i tumori sono generalmente benigni, la loro posizione e quantità possono compromettere la qualità della vita e portare a una mortalità precoce. In media, le persone con NF2 vivono fino all’età di 36 anni.
Nuove ricerche e prospettive per il 2024
Con il continuo progresso della ricerca, nel 2024 sono emerse nuove prospettive per la gestione della neurofibromatosi. Studi recenti hanno rivelato opzioni terapeutiche innovative, incluse terapie mirate e approcci genici, che potrebbero cambiare il modo in cui affrontiamo questa condizione. Ad esempio, i ricercatori stanno esaminando l’uso di farmaci che inibiscono specifici percorsi molecolari coinvolti nella crescita dei neurofibromi, mostrando risultati promettenti in studi clinici preliminari.
Inoltre, è stato evidenziato l’importanza di un approccio multidisciplinare nella gestione della neurofibromatosi, coinvolgendo neurologi, oncologi, specialisti della pelle e psicologi per fornire un supporto completo ai pazienti. Questo approccio non solo si concentra sui trattamenti fisici, ma anche sul benessere psicologico, che è cruciale per migliorare la qualità della vita dei pazienti.
Infine, la sensibilizzazione e l’educazione della comunità continuano a essere una priorità, poiché la comprensione della neurofibromatosi può portare a diagnosi più rapide e a migliori risultati per i pazienti. Con una continua ricerca e sviluppo, si spera che nel prossimo futuro possano emergere ulteriori terapie e risorse per migliorare la vita di chi vive con questa condizione.