La sindrome di Marfan è una condizione genetica che influisce sui tessuti connettivi, strutture fondamentali che forniscono supporto a vari organi e tessuti del corpo umano. Questa sindrome può interessare diverse parti del corpo, come il cuore, i vasi sanguigni, i polmoni, la pelle, le ossa, le articolazioni e gli occhi, portando a una vasta gamma di sintomi e complicazioni.
Gli effetti della sindrome di Marfan possono variare da lievi a gravi, con le complicazioni più serie che coinvolgono il cuore e l’aorta. È importante notare che, nonostante le sfide fisiche, questa condizione non influisce sulle capacità cognitive dell’individuo. La sindrome di Marfan è principalmente ereditaria, colpendo circa 1 persona su 5.000.
Attualmente, non esiste una cura definitiva per la sindrome di Marfan, ma un trattamento adeguato può migliorare significativamente la qualità della vita e prevenire complicazioni potenzialmente letali. Con un intervento tempestivo, una persona affetta può avere un’aspettativa di vita simile a quella di chi non ha la condizione.
Fatti Veloci Sulla Sindrome di Marfan
- La sindrome di Marfan è una condizione genetica che può causare una grande varietà di problemi cardiaci, oculari e scheletrici.
- I sintomi includono spesso braccia e dita insolitamente lunghe, altezza avanzata e possibili lacrime nell’aorta, che possono diventare evidenti anche in età adulta.
- La condizione è causata da anomalie in un gene che rinforza il tessuto connettivo.
- Non esiste una cura per la sindrome di Marfan, ma i sintomi possono essere gestiti e alleviati attraverso un’adeguata terapia.
- Una persona con sindrome di Marfan può avere un’aspettativa di vita normale se vengono adottate le giuste misure preventive.
Sintomi
Poiché la sindrome di Marfan può colpire varie parti del corpo, i sintomi possono differire significativamente da un individuo all’altro. Tuttavia, molti pazienti presentano una costituzione fisica caratteristica, con arti lunghi e talvolta deformità nel sistema scheletrico.
Uno dei segni più comuni è il rigonfiamento e le lacrime nell’aorta, il grande vaso sanguigno che trasporta il sangue dal cuore. I sintomi possono manifestarsi in modi diversi, influenzando il sistema scheletrico, gli occhi e il sistema cardiovascolare. In alcune persone, i sintomi sono lievi e limitati a specifiche aree del corpo; in altri, possono essere gravi e coinvolgere più sistemi. È importante notare che i sintomi tendono a peggiorare con l’età.
Segni e sintomi che possono apparire nel sistema scheletrico includono:
- Arti lunghi con polsi sottili e fragili.
- Spalle curve.
- Dita delle mani e dei piedi molto lunghe e sottili.
- Deformità dello sterno, che può sporgere o rientrare.
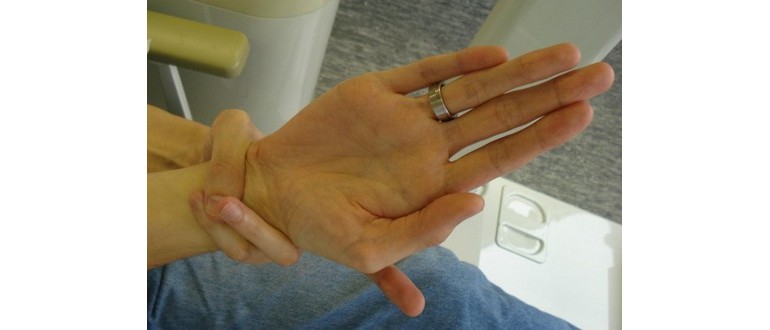
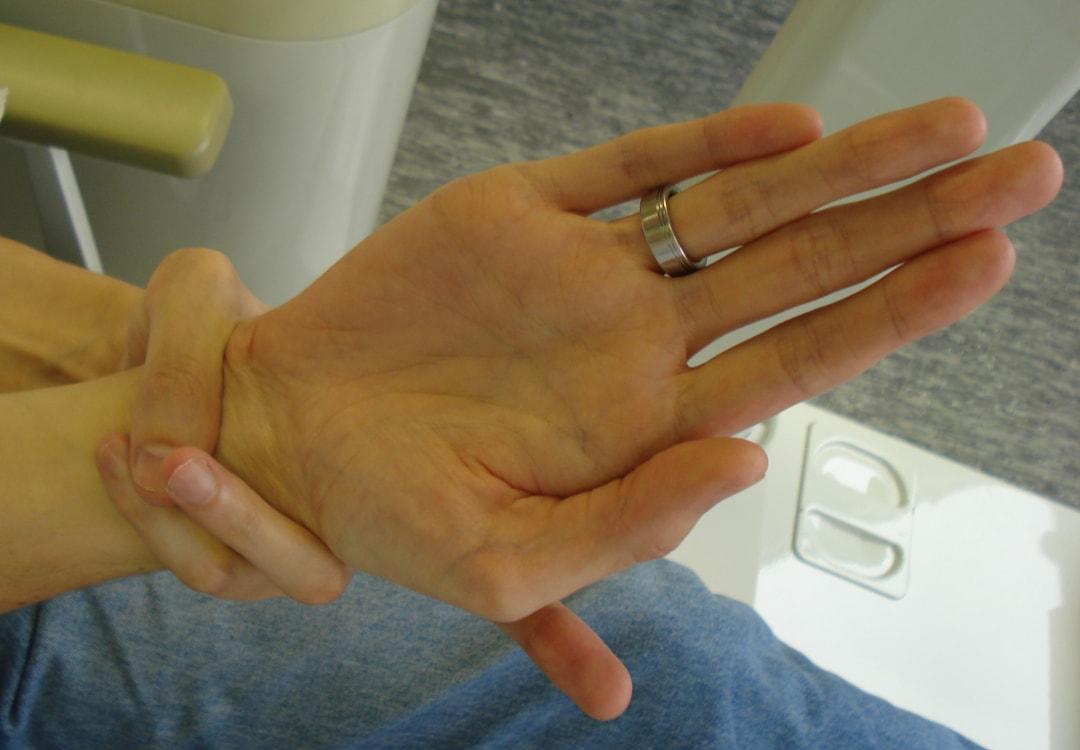
- Giunti estremamente flessibili.
- Volto lungo e stretto.
- Jawline piccola che può influenzare la pronuncia.
- Denti sovraffollati.
- Corpo snello e più alto della media.
- Piedi piatti.
- Arcata dentale alta che può causare difficoltà nel parlare.
- Smagliature cutanee non correlate a gravidanza o aumento di peso.
- Dolore articolare, osseo e muscolare.
Gli arti lunghi tipici della sindrome di Marfan possono far sì che la lunghezza delle braccia superi l’altezza dell’individuo. Inoltre, vi è un rischio aumentato di sviluppare scoliosi, spondilolistesi e ectasia durale, quest’ultima caratterizzata dall’allargamento del sacco durale che circonda il midollo spinale.
I problemi oculari associati alla sindrome di Marfan possono includere:
- Miopia.
- Astigmatismo.
- Dislocazione della lente.
- Retina distaccata.
Inoltre, i pazienti possono sviluppare glaucoma precoce e cataratta.
Le irregolarità aortiche possono comportare seri problemi cardiaci, tra cui:
- Affaticamento.
- Difficoltà respiratorie.
- Palpitazioni o soffi cardiaci.
- Angina, con dolore al petto che si irradia alla schiena, spalla o braccio.
- Prolasso delle valvole cardiache, inclusa l’aorta.
- Aorta dilatata.
- Aneurisma aortico.
È possibile che i sintomi della sindrome di Marfan non si manifestino fino all’età adulta.
Le Cause
La sindrome di Marfan è causata da mutazioni genetiche trasmesse attraverso le famiglie o da mutazioni spontanee. La maggior parte delle persone con questa sindrome eredita il gene difettoso da un genitore, e ogni genitore affetto ha una probabilità del 50% di trasmettere la condizione ai propri figli. Tuttavia, circa il 25% dei pazienti non ha un genitore con la sindrome, ma sviluppa la condizione a causa di una mutazione casuale.
Il gene responsabile della sindrome di Marfan è chiamato FBN1, il quale produce una proteina nota come Fibrillina-1, essenziale per conferire elasticità e forza ai tessuti connettivi. La mancanza di questa proteina compromette la capacità del tessuto connettivo di sostenere adeguatamente gli organi e le strutture corporee.
Una mutazione nel gene FBN1 interferisce con la produzione di Fibrillina-1, aumentando i livelli di alcune proteine infiammatorie, il che può portare a infiammazione e cicatrici nei tessuti connettivi.
Diagnosi
La diagnosi della sindrome di Marfan può risultare complicata a causa della varietà di sintomi. Un medico esperto procederà con un’accurata anamnesi familiare e personale del paziente, esaminando la storia clinica e i sintomi fisici.
Le valutazioni includeranno:
- Domande sulla storia familiare e medica del paziente.
- Ascolto del cuore del paziente.
- Controllo della pelle per le smagliature.
- Osservazione delle proporzioni delle braccia, delle gambe, delle dita e dei piedi del paziente.
È importante escludere altre condizioni con sintomi simili, come la sindrome di Ehlers-Danlos o la sindrome di Beals. La diagnosi nei bambini può essere difficile fino all’adolescenza, quando i segni diventano più evidenti.
Per confermare la diagnosi, il medico può utilizzare vari test, tra cui:
- Ecocardiogramma per valutare lo stato del cuore, delle valvole e dell’aorta.
- Elettrocardiogramma (ECG) per monitorare il ritmo e la frequenza cardiaca.
- Esame con lampada a fessura per verificare la presenza di lenti lussate.
- Scansioni TC o MRI per controllare la parte inferiore della schiena per segni di ectasia durale.
- Test genetici, se necessario, per confermare la diagnosi.
Trattamento
Sebbene non esista una cura per la sindrome di Marfan, ci sono diverse opzioni di trattamento per alleviare i sintomi e ridurre il rischio di complicazioni. Un piano di trattamento personalizzato sarà sviluppato dal medico in base ai sistemi corporei coinvolti.
Un monitoraggio regolare è cruciale per prevenire problemi ossei, articolari e tissutali, identificando tempestivamente eventuali alterazioni nella colonna vertebrale o nello sterno, soprattutto durante la crescita nei bambini.
Il trattamento tardivo può compromettere la funzione cardiaca e polmonare. In alcuni casi, potrebbe essere necessario l’uso di un tutore ortopedico o persino un intervento chirurgico.
Controlli ocularistici regolari sono essenziali per garantire che eventuali problemi visivi siano individuati e trattati tempestivamente. Secondo la National Marfan Foundation, molti problemi visivi possono essere corretti con occhiali o lenti, mentre altri potrebbero richiedere interventi chirurgici correttivi.
Un monitoraggio attento della salute cardiovascolare è fondamentale per prevenire complicazioni. I beta-bloccanti possono essere prescritti per gestire i problemi alle valvole cardiache e per ridurre la pressione all’interno dell’aorta. In alcuni casi, sarà necessario un intervento chirurgico per riparare l’aorta o sostituire una valvola cardiaca. Un intervento tempestivo è essenziale per prevenire gravi complicazioni, come la dissezione aortica.
La Fondazione Marfan raccomanda a tutti i pazienti con problemi cardiaci di indossare un braccialetto identificativo medico e di ricorrere immediatamente a cure ospedaliere in caso di dolore toracico, alla schiena o all’addome.
In alcuni casi, l’ectasia durale può influenzare il sistema nervoso centrale, richiedendo l’uso di farmaci per gestire il dolore.
Prospettive
Le persone con sindrome di Marfan sono generalmente sconsigliate dalla partecipazione a sport di contatto ad alta intensità. Tuttavia, attività come il bowling o il golf, che non comportano collisioni fisiche, sono generalmente sicure. È fondamentale consultare un medico per determinare quali sport siano adatti e sicuri.
Josephine Grima, PhD, Chief Science Officer della Fondazione Marfan, afferma: «Abbiamo fatto enormi progressi nella comprensione e nel trattamento della sindrome di Marfan. Di conseguenza, le persone affette possono condurre una vita normale se vengono diagnosticate e ricevono le cure mediche appropriate».
Per assistenza, supporto e informazioni sulla ricerca, visitare il sito web della Fondazione Marfan.
Nuove Ricerche e Approcci nel 2024
Negli ultimi anni, la ricerca sulla sindrome di Marfan ha fatto progressi significativi. Molti studi recenti si sono concentrati sull’identificazione precoce della condizione e sull’implementazione di strategie di trattamento personalizzate. Ad esempio, ricerche pubblicate nel 2024 hanno evidenziato l’importanza della telemedicina per il monitoraggio continuo dei pazienti, consentendo ai medici di intervenire rapidamente in caso di cambiamenti nei sintomi o nelle condizioni fisiche.
Inoltre, i ricercatori stanno esplorando nuovi approcci farmacologici per migliorare la qualità della vita dei pazienti, come l’uso di farmaci anti-infiammatori che possono ridurre i sintomi associati alla sindrome. La genetica continua a giocare un ruolo cruciale, con studi che cercano di meglio comprendere il legame tra mutazioni specifiche nel gene FBN1 e l’insorgenza di complicazioni cardiovascolari.
Infine, le associazioni di pazienti stanno svolgendo un ruolo chiave nel connettere i malati con le informazioni più recenti e nel promuovere la consapevolezza della sindrome di Marfan, incoraggiando una diagnosi precoce e un trattamento adeguato per migliorare gli esiti a lungo termine.