La fibrosi cistica è una malattia ereditaria che colpisce decine di migliaia di persone negli Stati Uniti e in tutto il mondo. Attualmente non esiste una cura per questa condizione, ma una nuova ricerca propone un approccio terapeutico innovativo che potrebbe impedire alla malattia di progredire.
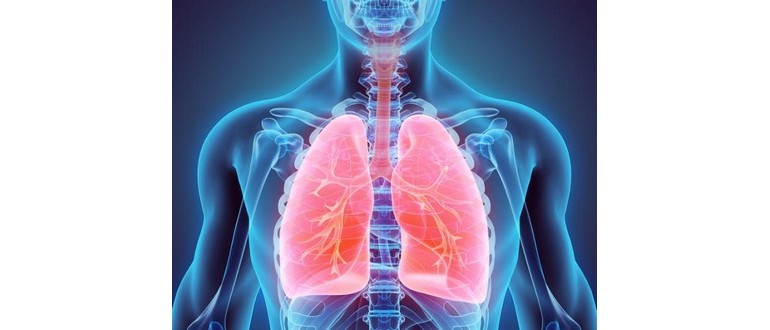
La fibrosi cistica (CF) è una malattia genetica delle ghiandole secretorie che colpisce circa 30.000 persone negli Stati Uniti e 70.000 persone in tutto il mondo. Le ghiandole secretorie sono responsabili della produzione di muco e sudore. Tuttavia, nella CF, queste secrezioni si accumulano nei polmoni, ostruendo le vie respiratorie e creando un ambiente favorevole alla crescita batterica. Di conseguenza, la condizione porta a gravi e ricorrenti infezioni polmonari.
Oltre ai polmoni, la CF influisce anche su altri organi vitali come il fegato e il pancreas, e può colpire l’intestino, i seni e gli organi riproduttivi del corpo.
Al momento, la fibrosi cistica è considerata incurabile. Tuttavia, un team internazionale di ricercatori della George Washington (GW) University di Washington, in collaborazione con l’Università di Perugia e l’Università di Roma (entrambe in Italia), ha recentemente scoperto un nuovo farmaco in grado di trattare e fermare la progressione della CF.
Lo studio, pubblicato su una rivista scientifica di rilievo, esamina gli effetti terapeutici della timosina alfa 1 (Tα1) sulla fibrosi cistica.
Tα1 è una versione sintetica di un polipeptide naturale isolato per la prima volta dal tessuto del timo, essenziale per il sistema immunitario.
Tα1 Riduce l’Infiammazione e Corregge i Difetti Genetici
La fibrosi cistica si verifica a causa di una mutazione nel codice genetico della proteina nota come regolatore di conduttanza transmembrana della fibrosi cistica (CFTR). Questa proteina è fondamentale per regolare l’attività del canale del cloruro, responsabile dell’equilibrio tra sale e acqua nei polmoni.
Nella CF, la mutazione conosciuta come «p.Phe508del» provoca una degradazione prematura della proteina CFTR, con conseguente scarsa permeabilità al cloro e infiammazione polmonare.
Nel nuovo studio, i ricercatori evidenziano che i complessi meccanismi patogenetici alla base della fibrosi cistica richiedono un approccio terapeutico combinato. Fino ad oggi, non è stato scoperto alcun farmaco singolo con molteplici effetti terapeutici per la CF.
Tuttavia, i risultati mostrano che Tα1 può correggere i difetti nel tessuto di topi con fibrosi cistica, oltre a migliorare le cellule di pazienti umani con la mutazione p.Phe508del.
Lo studio ha dimostrato che l’azione benefica di Tα1 è duplice: il farmaco riduce l’infiammazione tipica della CF e migliora l’attività, la maturazione e la stabilità del CFTR.
Tα1, commercialmente noto come «Zadaxin», è stato clinicamente approvato in più di 35 paesi per oltre 15 anni ed è frequentemente utilizzato per il trattamento di infezioni virali, disturbi da immunodeficienza e HIV.
Sebbene Zadaxin non sia attualmente disponibile negli Stati Uniti, i ricercatori affermano che il farmaco presenta «un eccellente profilo di sicurezza in clinica se usato come adiuvante o agente immunoterapeutico».
Allan L. Goldstein, Ph.D., coautore dello studio e professore emerito di biochimica e medicina molecolare presso la GW School of Medicine and Health Sciences, ha commentato l’importanza di questa ricerca:
«Attualmente esistono molteplici trattamenti per la fibrosi cistica, e mentre questi hanno migliorato significativamente l’aspettativa di vita, i pazienti continuano a vivere solo circa 40 anni. Nessun trattamento può affrontare autonomamente il difetto genetico alla base della fibrosi cistica e ridurre l’infiammazione che ne deriva.»
Nuove Prospettive per i Pazienti con Fibrosi Cistica nel 2024
Nel 2024, la ricerca sulla fibrosi cistica continua a evolversi, con studi recenti che esplorano l’efficacia di Tα1 e di altre molecole simili. Le evidenze suggeriscono che l’integrazione di terapie genetiche e immunologiche possa offrire ai pazienti un’opzione più robusta. Nuove statistiche mostrano che i pazienti trattati con approcci combinati presentano un miglioramento nell’efficienza respiratoria e nella qualità della vita.
Inoltre, recenti studi clinici stanno valutando la durata della risposta ai trattamenti, mostrando un potenziale per il prolungamento della vita e una riduzione degli effetti collaterali rispetto ai regimi terapeutici tradizionali. La comunità scientifica è ottimista riguardo a queste scoperte, che potrebbero portare a una vera rivoluzione nel trattamento della fibrosi cistica.
Insomma, la lotta contro la fibrosi cistica sta prendendo una nuova piega, e con l’emergere di terapie come Tα1, le aspettative per i pazienti sono più elevate che mai. È fondamentale restare informati e pronti ad adottare nuove strategie terapeutiche che promettono di cambiare il corso della malattia.