La sindrome di Apert è una rara malattia genetica caratterizzata dalla fusione prematura delle ossa del viso e del cranio durante lo sviluppo fetale.
Questa condizione provoca anomalie facciali e craniche, con ripercussioni significative sulla vista e sulla salute dentale. Inoltre, la sindrome di Apert può comportare anomalie nelle dita delle mani e dei piedi.
In questo articolo, esploreremo la sindrome di Apert, analizzando sintomi, trattamenti e prospettive per chi è affetto da questa condizione.
Qual è la sindrome di Apert?
La sindrome di Apert è una condizione in cui le ossa del cranio si uniscono prematuramente, influenzando la forma della testa e del viso.
Le persone nate con questa sindrome possono riscontrare problemi visivi e dentali dovuti alla conformazione anomala delle ossa facciali e craniche.
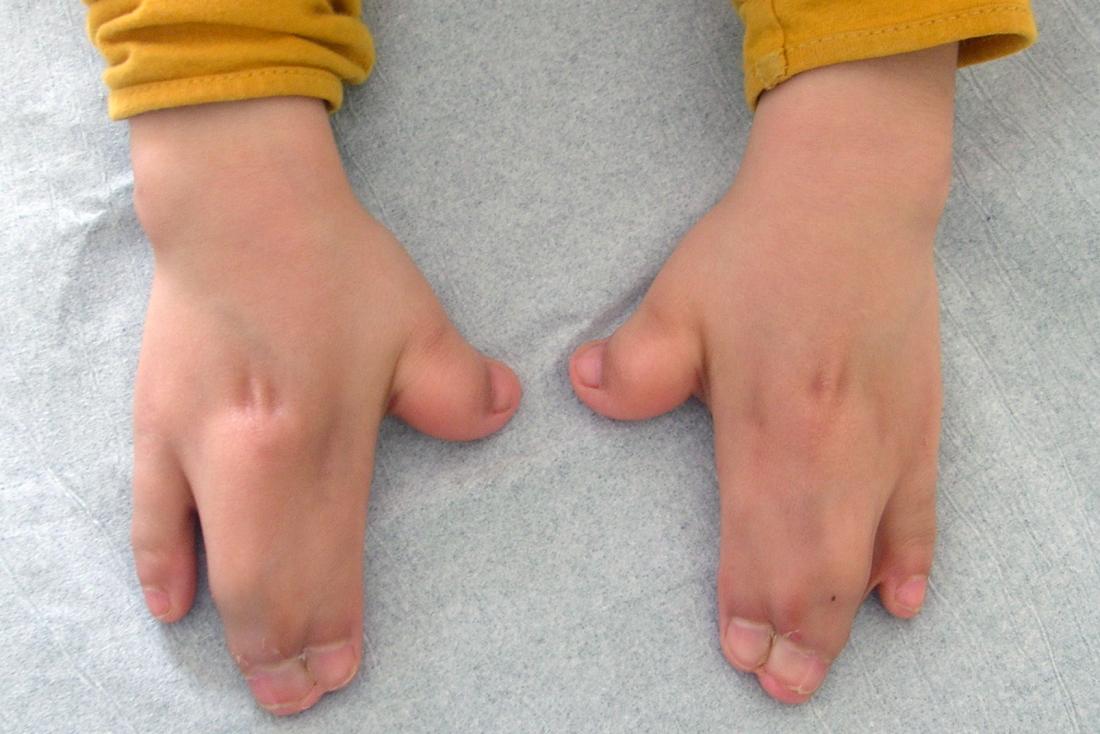
In molti casi, tre o più dita o dita dei piedi possono fondersi, una condizione nota come sindattilia.
La sindrome di Apert è ereditaria, ma spesso si presenta senza una storia familiare nota. Tuttavia, può essere trasmessa da un genitore.
Sintomi
I sintomi tipici della sindrome di Apert comprendono:
- un cranio a forma di cono, noto come turribrachicefalia
- una faccia incassata nel mezzo
- occhi sporgenti
- naso a becco
- mascella superiore sottosviluppata, che può causare affollamento dentale
Le anomalie cranio-facciali possono portare a problemi di salute e sviluppo. Senza correzione, è comune riscontrare problemi visivi a causa di orbite oculari poco profonde. Inoltre, la mascella superiore è spesso più piccola della media, provocando potenziali complicazioni dentali quando i denti del bambino iniziano a spuntare.
Il cranio di un individuo con sindrome di Apert è frequentemente più piccolo della norma, il che può esercitare pressione sul cervello in fase di sviluppo. Coloro che sono affetti possono avere un’intelligenza media o una compromissione intellettuale da lieve a moderata.
I bambini con questa sindrome presentano frequentemente dita palmate e piedi palmati. Generalmente, tre dita o dita dei piedi sono fuse, ma in alcuni casi, un’intera serie di dita può essere palmata. Raramente, possono essere presenti dita o dita dei piedi aggiuntivi.
Altri segni e sintomi associati alla sindrome di Apert includono:
- perdita dell’udito
- acne severa
- sudorazione eccessiva
- fusione delle ossa del midollo nel collo
- pelle grassa
- assenza di peli nelle sopracciglia
- ritardi nella crescita e nello sviluppo
- infezioni dell’orecchio ricorrenti
- palatoschisi
- disabilità intellettive da lievi a moderate
Le cause

La sindrome di Apert è causata da una mutazione genetica. Questa può verificarsi in bambini senza precedenti familiari di tale disturbo o può essere ereditata da un genitore.
Il gene coinvolto produce una proteina nota come recettore del fattore di crescita dei fibroblasti 2, che svolge ruoli cruciali nello sviluppo fetale, in particolare nel segnalare la formazione delle cellule ossee.
Quando si verifica una mutazione, il gene continua a inviare segnali a lungo, portando alla fusione prematura delle ossa del cranio, del viso, delle mani e dei piedi.
Queste mutazioni possono causare anche altri disturbi correlati, tra cui:
- Sindrome di Pfeiffer
- Sindrome di Crouzon
- Sindrome di Jackson-Weiss
Frequenza ed eredità
La sindrome di Apert è estremamente rara. Le statistiche esatte sul numero di affetti non sono note, con stime variabili tra le diverse fonti.
La National Library of Medicine degli Stati Uniti stima che colpisca 1 bambino su 65.000-88.000, mentre l’Organizzazione Nazionale per i Disturbi Rari (NORD) suggerisce che la cifra sia più vicina a 1 su 165.000-200.000 nascite.
La maggior parte dei casi di sindrome di Apert si verifica senza precedenti di malattia in famiglia. Tuttavia, il NORD indica che se un genitore ha la sindrome, c’è una probabilità del 50% che il bambino possa svilupparla, applicabile a ciascuna gravidanza.
Questa sindrome sembra colpire maschi e femmine in modo equo.
Diagnosi
La sindrome di Apert può essere diagnosticata alla nascita o in giovane età.
Per confermare la diagnosi, un medico esaminerà le anomalie ossee che interessano la testa, il viso, le mani e i piedi.
Possono essere richieste radiografie del cranio o TAC della testa per valutare la natura delle anomalie ossee. Anche i test genetici molecolari possono essere utili nella diagnosi.
Trattamento

Il trattamento della sindrome di Apert varia da persona a persona. I medici collaborano strettamente con il paziente e la sua famiglia per elaborare un piano di trattamento adeguato.
In alcuni casi, potrebbe essere raccomandato un intervento chirurgico per alleviare la pressione sul cervello.
Altre opzioni chirurgiche possono includere interventi per rimodellare le caratteristiche facciali o separare le dita palmati.
Un bambino con sindrome di Apert richiederà controlli regolari per tutta la vita. I medici monitoreranno eventuali complicazioni associate alla sindrome e suggeriranno trattamenti appropriati.
Le terapie comuni per le complicanze includono:
- correzione dei problemi visivi
- interventi per affrontare ritardi nello sviluppo e nella crescita
- procedure dentali per sistemare denti affollati
Prospettiva
La diagnosi precoce nei bambini aumenta le probabilità di un trattamento efficace per i sintomi e le complicanze associate alla sindrome di Apert.
In genere, questa sindrome non compromette l’aspettativa di vita, sebbene problemi cardiaci o altre condizioni associate possano portare a complicazioni.
Le prospettive a lungo termine per le persone affette da sindrome di Apert stanno migliorando grazie ai progressi della medicina moderna.
Le terapie possono spesso migliorare la qualità della vita degli individui con sindrome di Apert.
È fondamentale che un bambino con sindrome di Apert riceva cure cliniche continuative. Questo può includere supporto terapeutico per aiutare il bambino e la sua famiglia a gestire le sfide quotidiane associate a questa condizione.
Nuove Prospettive e Ricerche Recenti
Negli ultimi anni, la ricerca sulla sindrome di Apert ha compiuto passi da gigante. Studi recenti hanno evidenziato l’importanza di un approccio multidisciplinare nella gestione della sindrome, coinvolgendo specialisti in genetica, ortopedia, chirurgia plastica e terapia occupazionale. Questo approccio integrato ha dimostrato di migliorare notevolmente gli esiti clinici e la qualità della vita dei pazienti.
Inoltre, la terapia genica sta emergendo come una possibile via futura per affrontare le cause alla radice della sindrome. Nuove tecniche di editing genetico potrebbero, in teoria, correggere le mutazioni prima che causino le anomalie ossee, offrendo speranze per futuri trattamenti preventivi.
Statisticamente, l’accesso precoce a trattamenti e interventi chirurgici ha dimostrato di ridurre il rischio di complicazioni a lungo termine, rendendo l’importanza della diagnosi tempestiva ancora più evidente. Con una maggiore consapevolezza e un miglior accesso ai servizi sanitari, le prospettive per i bambini nati con sindrome di Apert sono più promettenti che mai.